
|
By: Daniel Wright

Illustration: Declan Wrede
FDA and Patent Timeline Mismatch
In its mission to protect citizens from faulty or fraudulent products, the Food and Drug Administration (FDA) requires you to submit substantial proof of both efficacy and safety of your new pharmaceutical or medical device before it can be legally sold in the United States. Collecting these data in clinical trials and the subsequent review process takes years, sometimes over a decade, and if there’s a patent involved, it’ll likely issue well before the FDA gives you the green light. This can seem more than a bit unfair since that patent term is supposed to provide the economic exclusivity needed to recuperate the fortune expended on drug development. Rest assured, the FDA is aware of this counterproductive interaction and offers a number of compensatory options. First, though, let’s take a look at the patent prosecution and FDA approval timelines for context.
MATCHING PACE: Patent Prosecution Timeline
The patent prosecution timeline has a few options depending on your target geographic market and whether you expedite your application, but shown here is the common path. Most applicants begin with a provisional application that yields an international patent application filing, usually under the Patent Cooperation Treaty (PCT), enabling the application’s entry into foreign jurisdictions. The duration of examination before each local patent office can vary dramatically, but in the U.S., this can take around one to three or more years. Following approval, your issued U.S. patent will remain enforceable for the patent’s term of twenty years starting from your first non-provisional filing date (as long as you keep paying the maintenance fees). Therefore, from start to finish, you’re looking at about four or more years to acquire your issued patent which will last for about sixteen years afterwards (note: the provisional patent application year is not counted in your 20 year patent term).

MATCHING PACE: Drug Discovery and Regulatory Timeline
Now, let’s review the other half of this discussion. The patent prosecution timeline can seem a mere blink of an eye in comparison to the duration that drug discovery and the regulatory process demand.
Preclinical research. The process begins in preclinical research. Here, chemists and biologists will decide on their biological target for the disease indication (e.g., which protein to inhibit to treat what illness), and design molecules to test for viable activity (e.g., does it actually inhibit the desired protein). Promising compounds will then be pushed into cell lines and animal models to probe for the drug’s in vivo efficacy, bioavailability, and toxicity. 
Investigational New Drug (IND) Application. With promising preclinical results in hand, you can then approach the FDA with an Investigational New Drug (IND) Application. In this document, you present all your evidence that suggests efficacy and safety in humans along with the parameters of your proposed clinical trials. The FDA will review it within thirty days for any unreasonable risk, demanding further experiments if needed, but, in bestowing approval, will authorize you to commence your clinical trials.
Clinical trials will likely be the longest period of this process because of the sheer scope of the undertaking. The mobilization of a sufficient patient population to explore short and long term effects in rigorous studies is not something that can be performed over a weekend, and for most, this operation will take about six to seven years, sometimes longer. New Drug Application (NDA). Ultimately, you will compile your (hopefully) successful clinical trial results along with relevant preclinical insights into a New Drug Application (NDA) that demonstrates the safety and efficacy of your new pharmaceutical towards one or more specified disease indications. The FDA has up to three years to return a final decision. If they find any aspect lacking, they may require additional studies to be performed; otherwise, with approval of the NDA comes your ability to finally sell your new pharmaceutical, perhaps only ten to fifteen years after you started. Note: Medical devices go through a very similar process, and if your drug is a generic of a compound previously approved by the FDA you can likely skip the clinical trials (we won’t be discussing those scenarios here). Patent Prosecution and FDA Timelines Side-by-Side
Having now explored both the patent prosecution and FDA timelines, let’s place them side by side. The exact alignment will depend upon when you chose to file your patent, but for many, the provisional application will be filed sometime near the IND filing due to the somewhat public nature of clinical trials and the concurrent demand for more open advertising and public relations. Accordingly, we end up with a dual timeline that looks something like this:
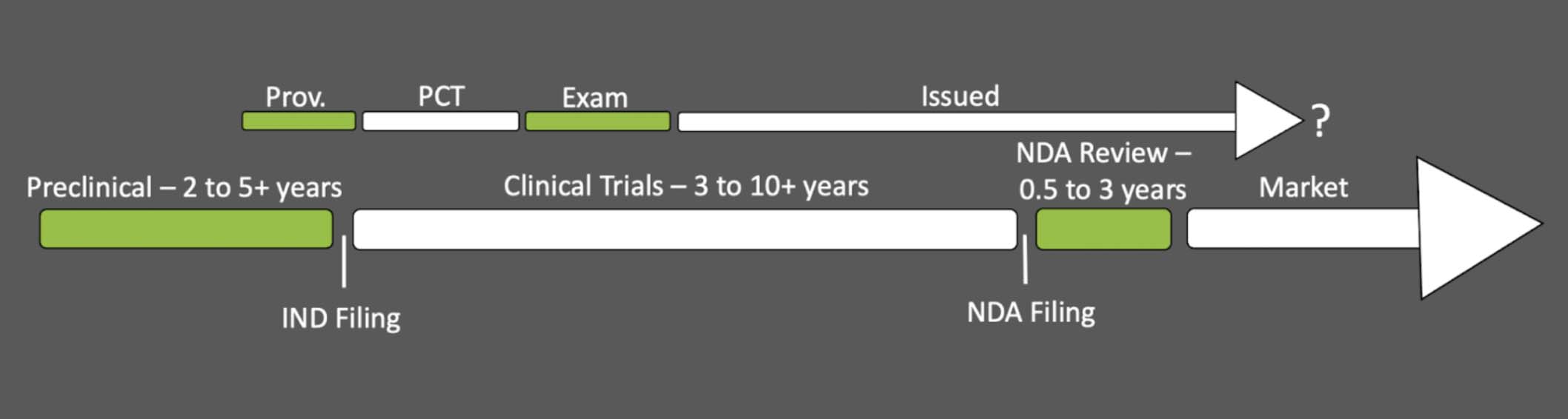
Very likely, you’ll acquire your issued patent sometime during clinical studies, well before you can enter the market. In the worst case scenario, your issued patent may have less than 5 years of life left when you can finally enter the market with your new drug. Let’s take a look at what the FDA offers as compensation for this less-than-ideal situation.
Options for Maximizing Your Exclusivity Window
In return for the extra regulatory hurdle and the potential lost patent protection time, the FDA offers the following three perks in certain conditions:
Patent Term Extension (PTE) means exactly what the words say: bonus term for your patent. Upon approval and at your timely request, the FDA will instruct the United States Patent and Trademark Office to append all of the time spent in NDA review and half the time between the IND and NDA filings to a single relevant patent. Up to a combined total of five years can be added, and the remaining lifetime of the chosen patent, after the addition, cannot exceed fourteen years. Furthermore, only the relevant claims enjoy the extension, so if your selected patent claims three discrete compounds separately, only the one towards the approved drug gets extended. The remaining two perks function independently of any related patents and instead only affect the FDA’s operations in your favor. New Chemical Entity (NCE) Exclusivity is an award to new “active moieties” of a five-year period in which the FDA will not accept any competing applications (usually an application for a generic of your drug). This means that, even if you have no patent on the drug, your successful FDA approval grants you a functionally similar limited monopoly on the market while the regulatory agency refuses to even initiate the process with competitors. Note that the FDA considers the “active moiety” of a drug to be the biologically active chemical entity regardless of any formulation, salt, or prodrug. If you’re working with any of the latter, you’ll be interested in what’s next. New Clinical Investigation Exclusivity is a three-year period in which the FDA will not approve competing applications. New Clinical Investigation Exclusivity is awarded to successful cases that involve a previously approved active moiety but had required new clinical studies. Applications that repurpose an old drug in a new formulation, prodrug, dosage, or towards a new disease indication can all qualify, but keep in mind that this perk is necessarily narrower than NCE Exclusivity. Assuming there’s no broader protection for your product, competitors could avoid your New Clinical Investigation Exclusivity by remaining with an older, unprotected formulation or marketing their generic only towards previously approved diseases. Start synchronizing your timelines…first steps to restful nights
The FDA certainly adds a whole other layer of complexity to the question (and we did not discuss here more niche perks such as those to pediatric or orphan drugs nor the nuanced tactics competitors can try to circumvent the various FDA exclusivities), but it’s comforting nonetheless that the agency is aware of the barrier its efforts present to the patent system and its motivating economic incentives. Certainly, you should discuss your filing strategy with your practitioner before committing to any plan, but you can sleep soundly knowing that some options remain even if your patent issues (or worse yet, is near expiration) well before your drug is on the market.
2 Comments
3/21/2023 06:39:45 pm
Thank you for noting that the FDA would direct the United States Patent and Trademark Office to include all of the time spent in NDA review and half of the time between the IND and NDA submissions in a single pertinent patent upon clearance and at your prompt request. My sister is employed in the pharmaceutical sector. To patent her items, she must speak with an expert. I'll assist her in finding the FDA regulatory consultant. Leave a Reply. |
Ashley Sloat, Ph.D.Startups have a unique set of patent strategy needs - so let this blog be a resource to you as you embark on your patent strategy journey. Archives
July 2024
Categories |
 RSS Feed
RSS Feed